WO2012053630A1 - 変異アンドロゲン受容体拮抗薬 - Google Patents
変異アンドロゲン受容体拮抗薬 Download PDFInfo
- Publication number
- WO2012053630A1 WO2012053630A1 PCT/JP2011/074261 JP2011074261W WO2012053630A1 WO 2012053630 A1 WO2012053630 A1 WO 2012053630A1 JP 2011074261 W JP2011074261 W JP 2011074261W WO 2012053630 A1 WO2012053630 A1 WO 2012053630A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carboxamide
- dimethylpiperazine
- cyanophenyl
- chloro
- compound
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/26—Androgens
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to a pharmaceutical composition, particularly an anticancer agent useful for the treatment of prostate cancer with androgen receptor mutation (AR mutation).
- AR mutation androgen receptor mutation
- Prostate cancer is one of the most common male malignancies in Western countries, accounting for about 20% of male cancer deaths. Moreover, since the frequency of prostate cancer increases with age, aging is an important generation factor. Prostate cancer research is considered to be of great significance for the future aging society. Many prostate cancers (about 80% or more) show androgen-dependent growth, so in patients with advanced prostate cancer, GnRH (Gonadotropin releasing hormone) analogs and androgen receptors (hereinafter abbreviated as ⁇ AR '') Antagonists are used in the treatment.
- GnRH Gonadotropin releasing hormone
- AWS Anti-androgen withdrawal syndrome
- an AR antagonist acts as an agonist
- the blood PSA Prostate Specific Antigen
- the PSA value may increase.
- the phenomenon in which the PSA level decreases by stopping the administration of the AR antagonist is AWS.
- AWS is found in approximately 30% of patients receiving AR antagonists, and AR mutations are thought to be the main cause.
- the AR antagonist currently used almost exclusively is Bicalutamide (hereinafter bicalutamide).
- bicalutamide Bicalutamide
- W741C mutant AR is detected, and bicalutamide acts as an agonist in PC-3, a prostate cancer cell that does not have AR, by introducing W741C mutant AR was reported.
- Flutamide Flutamide
- flutamide Flutamide
- hydroxyflutamide Hydroxyflutamide
- a novel AR antagonist that strongly antagonizes not only wild-type AR (normal AR) but also mutant AR (especially W741C mutant AR, ie bicalutamide-resistant mutant AR, and T877A mutant AR, ie, flutamide-resistant mutant AR) Is strongly desired.
- prostate cancer can be classified into the following stages (1) to (5) according to the progression of the disease state and the corresponding treatment method.
- the prostate cancer in (1) is called “Hormone-naive prostate cancer” and the prostate cancer in (2) is “Hormone sensitive prostate cancer ( HSPC)) ”.
- CRPC Basation resistant prostate cancer
- (3) falls under “chemo-naive CRPC”
- (5) falls under “chemo-failure CRPC” To do.
- Non-Patent Documents 1 to 4 can be referred to, and at least 44 types of AR mutations have been reported (Non-Patent Document 3).
- the AR mutation caused by the administration of bicalutamide, which is the first-line drug for the AR antagonist is the W741C mutant AR.
- Patent Document 1 In the formula, G is bicyclic or tricyclic aryl or heteroaryl, 5-membered heteroaryl, pyridyl, or substituted phenyl having a substituent different from the compound of the present invention. For details, refer to the publication. ) Patent Document 1 discloses the effectiveness of the compound of formula (a) against T877A mutation, L701H mutation, and H874Y mutation AR, but there is no disclosure or suggestion about the effectiveness against other types of mutation AR.
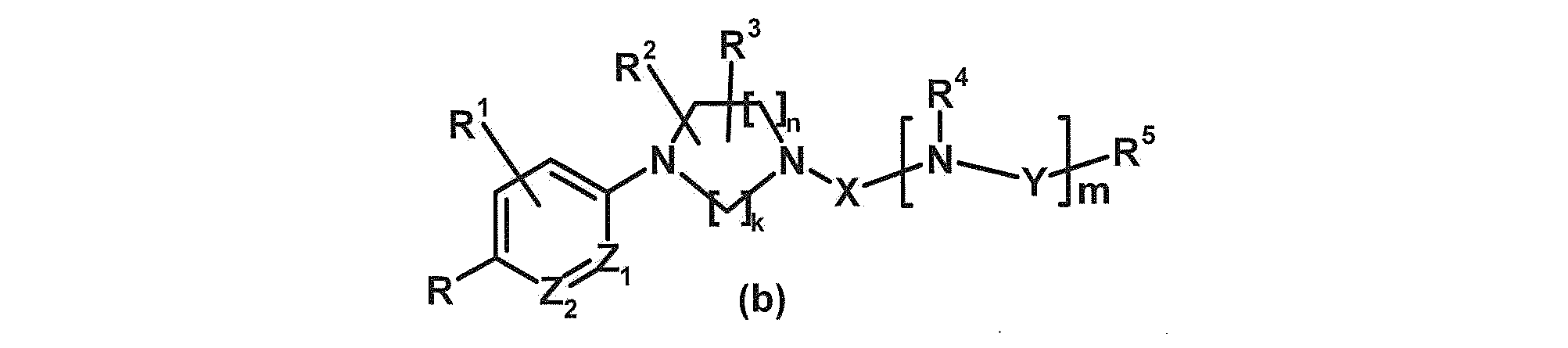
- Patent Document 2 a compound represented by the following formula (b) has been reported as an AR antagonist (Patent Document 2).
- R represents a cyano or nitro group
- Z 1 and Z 2 are the same or different and represent CH or a nitrogen atom
- X represents —C ( ⁇ O) —, —C ( ⁇ S) — or — S (O) 2 ⁇
- Patent Document 2 relates to an AR antagonist and a therapeutic agent for prostate cancer, benign prostatic hyperplasia, masculinosis, hirsutism, etc. in which AR is an exacerbation factor, but there is no disclosure or suggestion regarding mutant AR antagonism.
- Patent Document 3 a compound represented by the following formula (c) has been reported as an AR antagonist (Patent Document 3).
- Cy represents various substituted aryls or substituted heteroaryls.
- This patent document discloses the compound of Example 3-9 (Ex3-9) having the chemical structure shown above.
- this compound is referred to as Compound A (Compound A).
- Patent Document 3 relates to an AR antagonist and a therapeutic agent for prostate cancer, benign prostatic hyperplasia, masculinosis, hirsutism, etc. in which AR is an exacerbation factor, but there is no disclosure or suggestion regarding mutant AR antagonism.
- Patent Document 1 and Patent Documents 4 to 8 as compounds effective for mutant AR, when an AR antagonist effective for wild-type AR is invalidated by a specific AR mutation, Disclosure and suggestion of what kind of chemical structure conversion can be performed on an ineffective AR antagonist to obtain a compound having an effective anticancer action against the specific mutant AR, that is, a mutant AR antagonist No.
- An object of the present invention is to provide a novel therapeutic agent for prostate cancer with AR mutation, particularly bicalutamide-resistant or flutamide-resistant castration resistant prostate cancer (CRPC).
- CRPC castration resistant prostate cancer
- the present invention is useful for the treatment of prostate cancer with AR mutation, especially for bicalutamide resistance and flutamide resistance castration, which is newly found only in some of the compounds that are effective in the treatment of prostate cancer with AR mutation.
- the present invention relates to therapeutic use for resistant prostate cancer (CRPC).
- this invention contains the following. (1) (2R, 5S) -4- (3-chloro-4-cyanophenyl) -N- (2-cyclopropylpyrimidin-5-yl) -2,5-dimethylpiperazine-1-carboxamide, (2R, 5S) -4- (4-cyano-3-methoxyphenyl) -N- (2,6-dimethylpyrimidin-4-yl) -2,5-dimethylpiperazine-1-carboxamide, (2R, 5S) -4- (3-chloro-4-cyanophenyl) -N- (2-chloro-6-methylpyridin-4-yl) -2,5-dimethylpiperazine-1-carboxamide, (2R, 5S) -4- (3-chloro-4-cyanophenyl) -N- (2-methoxy-6-methylpyridin-4-yl) -2,5-dimethylpiperazine-1-carboxamide, (2R, 5S) -4- (3-chlor
- Examples of the compound that is an active ingredient of the pharmaceutical composition of the present invention include (2R, 5S) -4- (3-chloro-4-cyanophenyl) -N- (2-cyclopropylpyrimidin-5-yl)- 2,5-dimethylpiperazine-1-carboxamide.
- (2R, 5S) -4- (3-bromo-4-cyanophenyl) -N- (2-methoxypyrimidin-5-yl) -2,5-dimethylpiperazine-1-carboxamide is there.
- the present invention provides a pharmaceutical composition for the treatment of prostate cancer with AR mutation, particularly bicalutamide-resistant and flutamide-resistant castration resistant prostate cancer (CRPC).
- CRPC bicalutamide-resistant and flutamide-resistant castration resistant prostate cancer
- AR means androgen receptor.
- “Androgen receptor mutation” and “AR mutation” are amino acid sequence mutations caused by mutations in the gene encoding the protein of the androgen receptor due to various factors including drug administration. It is a phenomenon that exhibits different properties from the androgen receptor.
- the “AR mutation” is, for example, an “AR mutation” due to the use of an anticancer agent. In one embodiment, the “AR mutation” is due to the use of bicalutamide or flutamide.
- the “W741 mutation” is a kind of androgen receptor mutation found when a bicalutamide-administered patient reaches AWS, and is an AR mutation exhibiting a mutation in codon 741 of the natural AR gene.
- W741 mutation is “W741C mutation” or “W741L mutation”, and another embodiment is “W741C mutation”.
- the “W741C mutation” is an AR mutation in which the codon TGG coding for tryptophan is mutated to TGT coding for cysteine at codon 741 of the natural AR gene in the “W741 mutation”.
- the “W741L mutation” is an AR mutation in which the codon TGG encoding tryptophan is mutated to TTG encoding leucine at the codon 741 of the natural AR gene in the “W741 mutation”.
- the “T877 mutation” is a kind of androgen receptor mutation that occurs when a flutamide-administered patient reaches AWS, and is an AR mutation that exhibits a mutation in codon 877 of the natural AR gene.
- One embodiment of the “T877 mutation” is the “T877A mutation”.
- the “T877A mutation” is a T877 mutant AR in which the codon ACT encoding threonine is mutated to the codon GCT encoding alanine at the codon 877 of the natural AR gene among the above “T877 mutation”.
- “Bicalutamide resistance” is prostate cancer in which anti-androgen withdrawal syndrome (AWS) is observed due to administration of bicalutamide and the drug is ineffective. The main cause of bicalutamide resistance is the W741C mutation.
- “Flutamide resistance” is prostate cancer in which anti-androgen withdrawal syndrome (AWS) is observed due to administration of flutamide, and the drug is ineffective. The main cause of flutamide resistance is the T877A mutation.
- CRPC Ceration Resistant Prostate Cancer
- the compound which is an active ingredient of the pharmaceutical composition of the present invention other tautomers or optical isomers may exist depending on the type of substituent.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention may be described in only one form of these isomers, but these isomers are included, and the isomers are separated or mixed. Is also included.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention includes all these isomers.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention includes these pharmaceutically acceptable prodrugs.
- Pharmaceutically acceptable prodrugs are compounds having groups that can be converted to amino groups, OH, CO 2 H, etc. by solvolysis or under physiological conditions. Examples of groups that form prodrugs include those described in Prog. Med., 5, 2157-2161 (1985) and “Development of Pharmaceuticals” (Yodogawa Shoten, 1990), Volume 7, Molecular Design 163-198. Can be mentioned.
- the compound that is an active ingredient of the pharmaceutical composition of the present invention may form an acid addition salt or a salt with a base depending on the type of substituent, and such a salt is a pharmaceutically acceptable salt. To the extent they are included in the present invention.
- inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid
- organic acids such as lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, aspartic acid or glutamic acid, sodium, potassium, magnesium, calcium, aluminum, etc.
- Inorganic bases salts with organic bases such as methylamine, ethylamine, ethanolamine, lysine, ornithine, ammonium salts, and the like.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention and pharmaceutically acceptable salts thereof include various hydrates, solvates and polymorphic substances.
- the compounds that are the active ingredients of the pharmaceutical composition of the present invention and pharmaceutically acceptable salts thereof include compounds labeled with various radioactive or non-radioactive isotopes.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention and a pharmaceutically acceptable salt thereof can be obtained by applying various known synthetic methods using characteristics based on the basic skeleton or the type of substituent. Can be manufactured. In this case, depending on the type of functional group, it is effective in terms of production technology to replace the functional group with an appropriate protecting group (a group that can be easily converted into the functional group) at the raw material or intermediate stage. There is. Examples of such a functional group include an amino group, a hydroxyl group, and a carboxyl group, and examples of protective groups thereof include, for example, “Greene's Protective Groups in Organic Synthesis (No. 4) by PGM Wuts and Green (TW Greene)”.
- a desired compound can be obtained by introducing the protecting group and carrying out the reaction, and then removing the protecting group as necessary.
- the prodrug of the compound which is an active ingredient of the pharmaceutical composition of the present invention introduces a specific group at the stage from the raw material to the intermediate, or reacts further using the obtained compound, in the same manner as the protective group. It can be manufactured by doing.
- the reaction can be carried out by applying a method known by those skilled in the art, such as ordinary esterification, amidation, dehydration and the like.
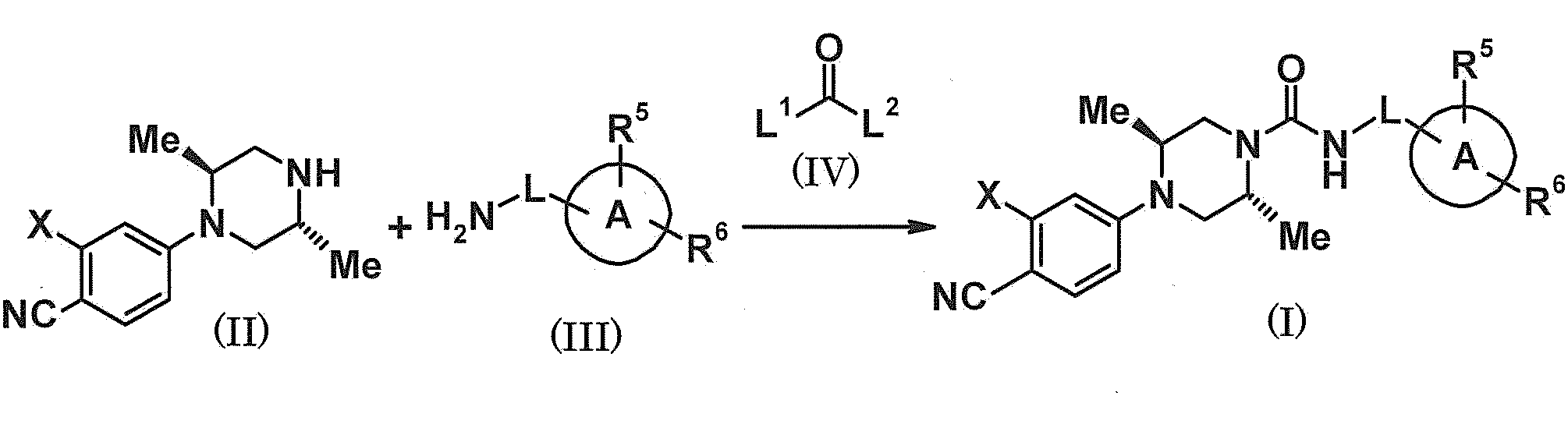
- the compound which is an active ingredient of the pharmaceutical composition of the present invention can be produced by the reaction of compound (II) and compound (III) and compound (IV) as an activated carbonic acid derivative serving as a C ⁇ O source.
- suitable carbonic acid derivatives include triphosgene, phenyl chlorocarbonate, N, N′-carbonyldiimidazole (CDI), and others that are obvious to those skilled in the art. (In the formula, symbols in the formula mean the following.
- X Cl, Br, methoxy, ethoxy or isopropyloxy
- L a bond or methylene
- Ring A phenyl, pyridyl, pyrimidinyl, indolyl, quinolyl, or quinoxalyl
- R 5 and R 6 the same or different from each other, —CO—N (CH 3 ) 2 , F, methoxy, methyl, ethyl, or cyclopropyl
- the compound (I) means any one of Example compounds Ex1 to 22 and Reference compound Ref1-1 to 1-3 described later.
- L 1 and L 2 represent a leaving group.
- solvent used here are not particularly limited, but aromatic hydrocarbons such as benzene, toluene or xylene, halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane (DCE) or chloroform, diethyl
- aromatic hydrocarbons such as benzene, toluene or xylene
- halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane (DCE) or chloroform
- diethyl examples include ethers such as ether, tetrahydrofuran (THF), dioxane, dimethoxyethane, N, N-dimethylformamide (DMF), dimethyl sulfoxide, ethyl acetate, acetonitrile or water, and mixtures thereof.
- the compound which is an active ingredient of the pharmaceutical composition of this invention can be manufactured by making the compound (VI) manufactured using the compound (II) and the compound (V) as a raw material react.
- the compound (II) and the compound (VI) can be produced by condensation.
- the compound (VI) can be produced by the rearrangement reaction in the reaction system after the compound (V) is azidated using diphenylphosphoryl azide (DPPA) or other azidating agent.
- DPPA diphenylphosphoryl azide
- the condensation reaction succeeding the rearrangement reaction in this production method is a method well known to those skilled in the art, and the reaction solvent, temperature and the like can be carried out in the same manner as in the above production method 1. Specifically, examples described later and the first production method of Patent Document 7 can be referred to.
- X 1 represents a leaving group
- P represents an amino-protecting group.
- Compound (II) can be produced by deprotecting the compound (VII) and compound (VIII) after substitution reaction.
- examples of the leaving group X 1 include halogen, methanesulfonyloxy, p-toluenesulfonyloxy group, and the like.
- the amino-protecting group P is, for example, benzyl or t-butoxycarbonyl group.
- compound (VII) and compound (VIII) are used in an equal amount or in excess, and the mixture is preferably used in a solvent inert to the reaction or in the absence of a solvent from cooling to heating under reflux. Is usually stirred at 0 to 80 ° C. for 0.1 hour to 5 days.
- solvent used here include, but are not limited to, aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as diethyl ether, tetrahydrofuran, dioxane and dimethoxyethane, dichloromethane and 1,2-dichloroethane.
- Halogenated hydrocarbons such as chloroform, N, N-dimethylformamide, dimethyl sulfoxide, ethyl acetate, acetonitrile, and mixtures thereof.
- the reaction is carried out in the presence of an organic base such as triethylamine, N, N-diisopropylethylamine or N-methylmorpholine, or an inorganic base such as potassium carbonate, sodium carbonate or potassium hydroxide. May be advantageous.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention is isolated and purified as a free compound, a salt thereof, a hydrate, a solvate or a crystalline polymorphic substance.
- the salt of the compound can also be produced by subjecting it to a conventional salt formation reaction. Isolation and purification are performed by applying ordinary chemical operations such as extraction, fractional crystallization, and various fractional chromatography. Various isomers can be produced by selecting an appropriate raw material compound, or can be separated by utilizing a difference in physicochemical properties between isomers.
- optical isomers can be obtained by general optical resolution of racemates (for example, fractional crystallization leading to diastereomeric salts with optically active bases or acids, chromatography using chiral columns, etc.). Further, it can also be produced from a suitable optically active raw material compound.
- the pharmaceutical composition of the present invention is prepared using one or more active ingredients and carriers, excipients, and other additives that are usually used for formulation.
- Administration is oral by tablet, pill, capsule, granule, powder, liquid, etc., or parenteral by injection, intravenous injection, intramuscular injection, suppository, transdermal agent, nasal agent, inhalant, etc. Either form may be sufficient.
- the dosage is appropriately determined according to the individual case, taking into account the symptoms, age of the subject, sex, etc. In general, in the case of oral administration, it is about 0.001 mg / kg to 100 mg / kg per day for an adult. This is administered once, or divided into 2 to 4 times.
- intravenously administered when intravenously administered depending on symptoms, it is usually administered once or several times a day in the range of 0.0001 mg / kg to 10 mg / kg per adult. In the case of inhalation, it is usually administered once or several times a day in the range of 0.0001 mg / kg to 1 mg / kg per adult.
- solid composition for oral administration tablets, powders, granules and the like are used.
- one or more active substances are present in at least one inert excipient such as lactose, mannitol, glucose, hydroxypropylcellulose, microcrystalline cellulose, starch, polyvinylpyrrolidone, metasilicate.
- the composition may contain an inert additive, for example, a lubricant such as magnesium stearate, a disintegrant such as sodium carboxymethyl starch, a solubilizer, and a solubilizing agent according to a conventional method.
- tablets or pills may be coated with a sugar coating or a gastric or enteric coating agent.
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, elixirs, etc., and include commonly used inert solvents such as purified water, ethanol, etc. .
- the composition may contain adjuvants such as solubilizers, wetting agents, and suspending agents, sweeteners, corrigents, fragrances, and preservatives.
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions.
- aqueous solvent include distilled water for injection and physiological saline.
- non-aqueous solvent include propylene glycol, polyethylene glycol, vegetable oil such as olive oil, alcohols such as ethanol, polysorbate 80 (trade name), and the like.
- Such compositions may further contain isotonic agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers, solubilizers, and solubilizing agents. These are sterilized by, for example, filtration through a bacteria-retaining filter, blending with a bactericide or irradiation. These can also be used by producing a sterile solid composition and dissolving and suspending it in sterile water or a sterile solvent for injection before use.
- a transmucosal agent such as an inhalant or a nasal agent
- a solid, liquid or semi-solid agent is used and can be produced according to a conventionally known method.
- excipients such as lactose and starch, and pH adjusters, preservatives, surfactants, lubricants, stabilizers, thickeners and the like may be added as appropriate.
- an appropriate device for inhalation or insufflation can be used.
- a known device such as a metered dose inhalation device or a nebulizer
- the compound is administered alone or as a powder in a formulated mixture or as a solution or suspension in combination with a pharmaceutically acceptable carrier. be able to.
- the dry powder inhaler or the like may be for single or multiple administration, and a dry powder or a powder-containing capsule can be used. Alternatively, it may be in the form of a pressurized aerosol spray using a suitable propellant, for example, a suitable gas such as chlorofluoroalkane, hydrofluoroalkane or carbon dioxide.
- a suitable propellant for example, a suitable gas such as chlorofluoroalkane, hydrofluoroalkane or carbon dioxide.
- the appropriate daily dose is about 0.001 to 100 mg / kg, preferably 0.1 to 30 mg / kg, more preferably 0.1 to 10 mg / kg per body weight. Or in 2 to 4 divided doses.
- the appropriate daily dose is about 0.0001 to 10 mg / kg per body weight, and is administered once to several times a day.
- a transmucosal agent about 0.001 to 100 mg / kg per body weight is administered once to several times a day. The dose is appropriately determined according to individual cases in consideration of symptoms, age, sex, and the like.
- the pharmaceutical composition of the present invention is 0.01 to 100% by weight, and in one embodiment, 0.01 to 50% by weight of the active ingredient. Or the compound or its salt which is the active ingredient of the pharmaceutical composition of this invention beyond it.
- the pharmaceutical composition of the present invention can be used in combination with other therapeutic agents or preventive agents effective for mutant AR prostate cancer.
- the drug a drug causing an AR mutation is excluded.
- the combination may be administered simultaneously, separately separately, or at desired time intervals.
- the simultaneous administration preparation may be a compounding agent or may be separately formulated.
- a compound is not limited to the compound as described in the following Example.
- the manufacturing method of a raw material compound is shown in a manufacture example, respectively, and the compound for comparing with an Example compound is shown as a reference example compound.
- the reference compound is a compound having an N-phenyl- (2R, 5S) -dimethylpiperazine structure, which is common to compounds that are active ingredients of the pharmaceutical composition of the present invention, but the antagonistic action against mutant AR is greatly attenuated. It is a compound.
- M represents [mol / L]
- ESI + represents an m / z value in mass spectrometry (ionization method ESI, (M + H) + unless otherwise specified)
- EI + represents EI [M] + .
- Example 1 4-[( ⁇ [(2R, 5S) -4- (3-Chloro-4-cyanophenyl) -2,5-dimethylpiperazin-1-yl] carbonyl ⁇ amino) methyl] benzoic acid (256 mg) and DMF (3 ml) was added to a mixture of dimethylamine hydrochloride (147 mg), 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (WSCHCl) (173 mg), 1-hydroxy- 1H-benzotriazole (HOBt) (41 mg) and triethylamine (0.251 ml) were added, and the mixture was stirred at room temperature for 18 hours.
- Example 6 To a mixture of 2-methoxypyrimidin-5-amine (120 mg) and pyridine (4 ml) was added phenyl chlorocarbonate (0.132 ml) under ice cooling, and the mixture was stirred at room temperature for 1.5 hours. Subsequently, a mixture of 2-chloro-4-[(2S, 5R) -2,5-dimethylpiperazin-1-yl] benzonitrile (200 mg) and pyridine (3 ml) was added, and the mixture was stirred at 100 ° C. for 1 hour. The reaction mixture was allowed to cool and then concentrated under reduced pressure. Toluene (15 ml x 2) was added to the residue and azeotroped.
- Example 16 Triethylamine (0.184 ml) and DPPA (0.208 ml) were added to a mixture of 2-chloro-6-methylisonicotinic acid (151 mg) and ethyl acetate (3 ml), and the mixture was stirred at room temperature for 2.5 hours. Toluene (15 ml) was added to the reaction solution, washed with a saturated aqueous solution of sodium hydrogen carbonate, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 22 (2R, 5S) -4- (3-chloro-4-cyanophenyl) -N- (2-cyclopropylpyrimidin-5-yl) -2,5 which is the compound of Example 3-9 of Patent Document 3 -Dimethylpiperazine-1-carboxamide (compound A) (250 mg) was crystallized with 50% aqueous acetonitrile (6 ml) at room temperature to give (2R, 5S) -4- (3-chloro-4-cyanophenyl) -N Crystals of-(2-cyclopropylpyrimidin-5-yl) -2,5-dimethylpiperazine-1-carboxamide monohydrate (111 mg) (44%) were obtained.
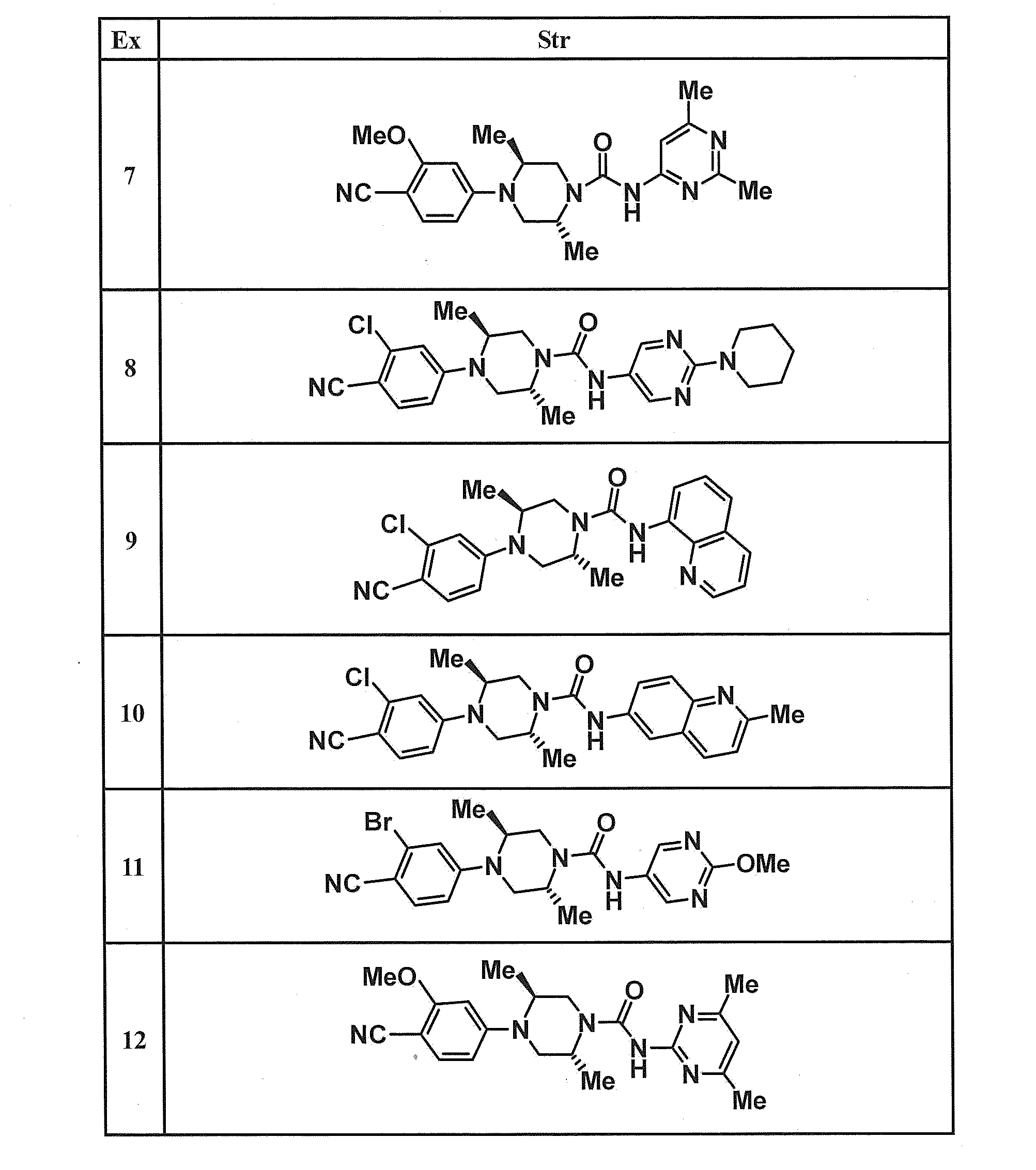
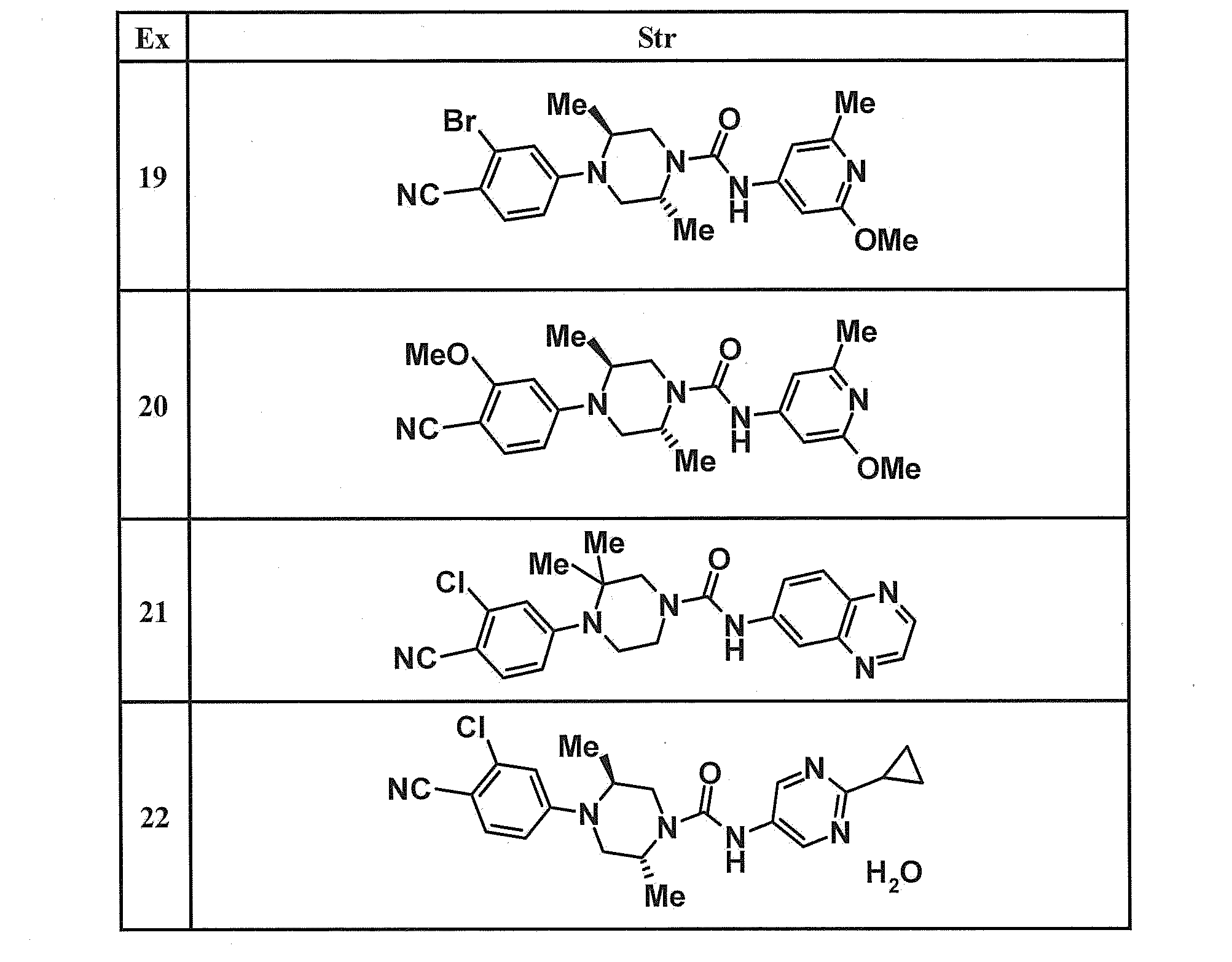
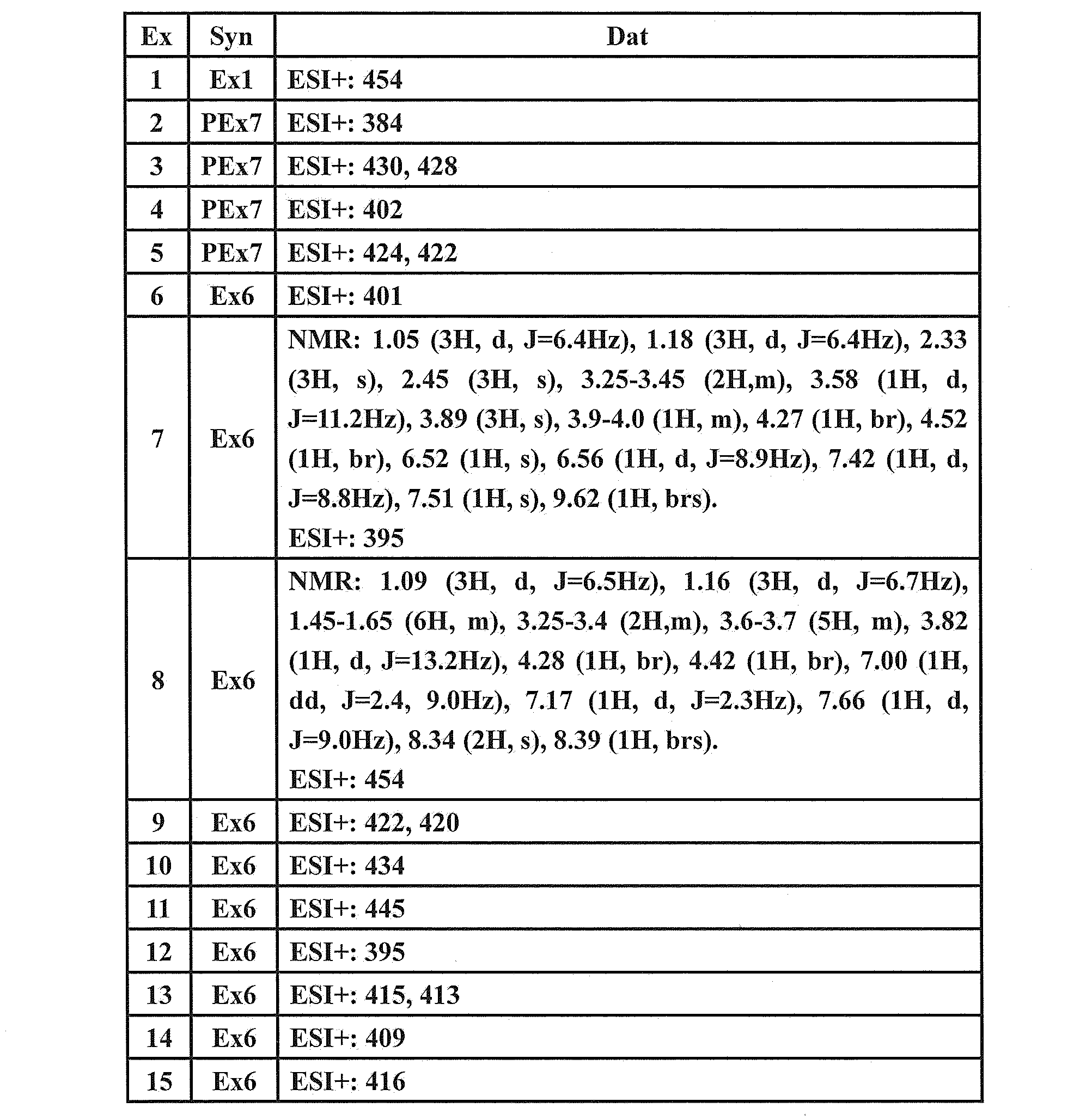
- Example compounds and Reference Example compounds shown in the table below were produced using corresponding raw materials.
- the structural formulas, physicochemical data, and production methods of the Example compounds and Reference Example compounds are shown in the following table.
- Test Example 1 Human W741C and T877A Mutant AR Transcriptional Activation Inhibitory Effect
- a W741C or T877A mutant AR expression vector pSG5-W741C-hAR or pcDNA3.1-T877A-hAR
- W741C and T877A mutant AR stably expressing cells were obtained. These cells were also transfected with a luciferase reporter vector so that luciferase was expressed when AR was activated, and human W741C and T877A mutant AR stably expressing cells were obtained.
- IC 50 values were calculated by Sigmoid-Emax model nonlinear regression analysis.
- test compounds in the above test bicalutamide, hydroxyflutamide, compound which is an active ingredient of the present invention (compound A (Compound A, Patent Document 3, Examples 3-9), Examples 7 and 8) , 11, 16, 18, 19, and 22), and Reference Examples 1-1 to 1-3, transcription to the respective receptors of wild-type AR (Wild), W741C mutant AR, and T877A mutant AR Activation inhibitory action was evaluated.
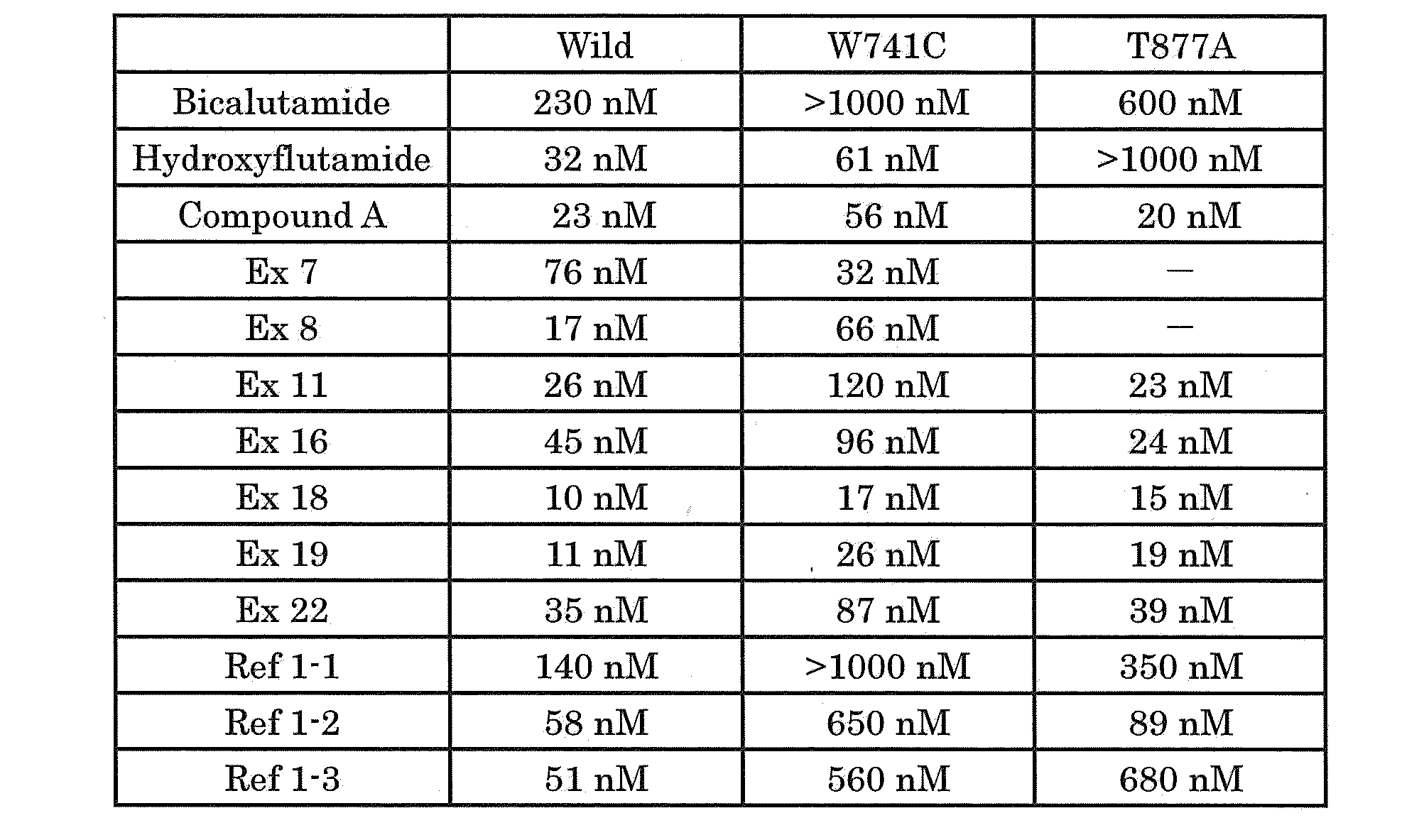
- the receptor inhibitory action of each test compound is calculated as an IC 50 value, and the value is shown in the table below. “-” Means not implemented.
- An IC 50 value of> 1000 nM in the table means that no transcription activation inhibitory effect is found and AR antagonistic activity is almost ineffective.
- Bicalutamide is an existing AR antagonist, and the transcription activation inhibitory effect having the W741C mutation, which is characteristic of bicalutamide resistance, was not found, and it was confirmed that the antagonistic activity was almost ineffective against the W741C mutant AR receptor.
- Hydroxyflutamide is an active metabolite of flutamide, and it was confirmed that antagonistic activity was almost ineffective against the AR receptor having the A877A mutation, which is characteristic of flutamide resistance.
- Compound A Compound A
- compounds of Examples 7, 8, 11, 16, 18, 19, and 22, which are compounds that are active ingredients of the pharmaceutical composition of the present invention are wild-type AR, W741C mutant AR, and A877A. Transcriptional activation inhibitory action was observed for any of the mutant ARs, and it was confirmed to have AR antagonistic activity.
- the compounds of Reference Examples 1-1, 1-2, and 1-3 have a transcriptional activation inhibitory action on wild-type AR and wild-type AR antagonistic activity, but compound A (Compound A), Compared with the compounds of Examples 7, 8, 11, 16, 18, 19, and 22, the transcriptional activation inhibitory action on W741C and / or T877A mutant AR, or both, is greatly attenuated, and therefore the mutant AR antagonistic activity is large. It was confirmed that it was attenuated.
- Example 22 (Ex 22) is a monohydrate of Compound A and is not disclosed or suggested in Patent Document 3.
- Test Example 2 Antitumor activity in KUCaP tumor-bearing mice
- KUCaP is a human prostate cancer cell established at Kyoto University (Cancer Res 2005; 65: 9611-9616), has a W741C mutant AR, in vivo (mouse ).
- This cell fragment was transplanted subcutaneously in the back of male SCID mice, and when the tumor volume reached approximately 200-400 mm 3 , the cells were divided into groups (5 per group) so that the tumor volume was equal in each group.
- Administration of the test compound was started.
- the test compound was dissolved in 25% propylene glycol / 25% Tween 80/50% purified water and orally administered at 5 mg / kg / 10 mL twice a day (10 mg / kg / day) for 14 days.
- Inhibition rate [%] 100 ⁇ ⁇ 1-((Tumor volume on day 14 of test compound group ⁇ tumor volume on day of administration of test compound group)] / [(Tumor volume on day 14 of control group ⁇ control) Tumor volume on day of administration of group)] ⁇
- Test Example 2 several compounds that are active ingredients of the pharmaceutical composition of the present invention were evaluated.
- the following table shows the inhibition rate [%].
- Ex means an Example compound number.
- Test Example 3 Evaluation of human W741C and T877A mutant androgen receptor (AR) binding activity CHO-K1 cells are transfected with a W741C or T877A mutant AR expression vector (pSG5-W741C-hAR or pcDNA3.1-T877A-hAR). Thus, human W741C and T877A mutant AR transient forced expression cells were obtained. The cells were seeded on a 24-well plate at 5 ⁇ 10 4 cells / well and cultured overnight at 37 ° C. The medium was removed, a test compound diluted with a medium supplemented with DCC-FBS and [ 3 H] DHT were added, and the mixture was cultured at 37 ° C. for 4 hours.
- a test compound diluted with a medium supplemented with DCC-FBS and [ 3 H] DHT were added, and the mixture was cultured at 37 ° C. for 4 hours.
- lysis buffer was added to lyse the cells, and the radioactivity of the supernatant was measured. From this radioactivity value, the IC 50 of the inhibitory activity of the test compound for the specific binding of [ 3 H] DHT was determined.
- Test Example 4 Effect on proliferation of W741C mutant AR-expressing human prostate cancer cell line LNCaP (W741C-hAR-LNCaP) 1) Acquisition of W741C-hAR-LNCaP W741C-hAR-LNCaP was obtained by transfecting L74aC cells, a human prostate cancer cell line, with a W741C mutant AR expression vector (pSG5-W741C-hAR).
- W741C-hAR-LNCaP growth inhibitory action 96-well plate coated with Poly-L-lysine W741C-hAR-LNCaP cells are seeded at 5 ⁇ 10 3 cell / well and cultured for 1 day, and contain DCC-FBS Both the test compound (or solvent) diluted with the medium and DHT (1 nM) were added and further cultured. Seven days later, the protein amount in each well was measured using the sulforhodamine B method. The inhibition rate (%) was calculated by the following formula.
- Inhibition rate (%) 100 [(IB)-(X-B)] / (IB)
- I Protein mass when only 1 nM DHT is added
- B Protein mass when only solvent is added
- X Protein mass when this compound and 1 nM DHT are added simultaneously
- references 1 and 2 can be referred to.
- Reference 1 Skehan P, Storeng R, Scudiero D et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82: 1107-1112.
- Reference 2 Papazisis KT, Geromichalos GD, Dimitriadis KA et al. Optimization of the sulforhodamine B colorimetric assay. J Immunol Methods 1997; 208: 151-158.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention has a W741C and T877A mutant AR transcription activation inhibitory action, and a human prostate cancer cell having the W741C mutant AR It was confirmed to have an antitumor effect in mice transplanted with. Therefore, it was shown that the compound which is an active ingredient of the pharmaceutical composition of the present invention is useful for the treatment of AR-related diseases with AR mutation, particularly prostate cancer with AR mutation.
- the compound which is an active ingredient of the pharmaceutical composition of the present invention or a pharmaceutically acceptable salt thereof exhibits excellent antagonistic activity against the mutant AR, and is excellent in an anti-tumor tumor in a prostate cancer animal model with the AR mutation Since the action was recognized, the present invention is useful as a therapeutic agent for prostate cancer with AR mutation.
Abstract
Description
前立腺癌の多く(約80%以上)は、アンドロゲン依存性増殖を示すことから、進行性前立腺癌患者において、GnRH(Gonadotropin releasing hormone)アナログやアンドロゲン受容体(Androgen receptor、以下「AR」と略す)拮抗薬がその治療に用いられている。投与初期は前立腺癌の増殖を強力に阻害するが、数年の内にその治療に再燃することが本疾患の大きな問題となっている。その再燃の一つとして、AWS(Anti-androgen withdrawal syndrome)という現象(AR拮抗薬がアゴニストとして働く現象)が認められている(J. Cell. Biochem 2007, 91, 3-12)。すなわち、GnRHアナログとAR拮抗薬を併用している前立腺癌患者において、投与開始から一定期間は前立腺癌の指標である血中PSA(Prostate Specific Antigen)値が低下しているが、投与を続けているとPSA値が増加することがある。その時にAR拮抗薬の投薬を中止することにより、PSA値が低下する現象がAWSである。AWSは、AR拮抗薬投与患者の約30%で認められており、AR変異が主原因と考えられている。現在、ほぼ独占的に使用されているAR拮抗薬は、Bicalutamide(以下、ビカルタミド)である。ビカルタミドが無効になった前立腺癌患者において、W741C変異ARが検出され、また、ARを有しない前立腺癌細胞であるPC-3において、W741C変異ARを導入することにより、ビカルタミドがアゴニストとして作用することが報告された。これらのことから、ビカルタミドのAWSには、W741C変異ARが関与していることが示唆された。また、ビカルタミド以外のAR拮抗薬として、Flutamide(以下、フルタミド)があるが、肝臓および消化管障害ならびに1日3回投与のコンプライアンスの悪さにより、現在ほとんど使用されていない。このフルタミドの活性代謝物であるHydroxyflutamide(ヒドロキシフルタミド)は、T877A変異ARにおいてアゴニストとして作用することが知られている。
(1) 進行性前立腺癌と診断。
(2)ホルモン療法を開始:GnRHアナログ及びAR拮抗薬(通常はビカルタミド)が用いられる。
(3)AR拮抗薬(通常はビカルタミド)に耐性を示し、当該AR拮抗薬が無効になる。
(4)化学療法剤ドセタキセルの使用開始。
(5)ドセタキセルに対しても耐性を示し、無効になる。
上記ステージにおいて、(1)における前立腺癌は「ホルモン療法剤未処置前立腺癌(Hormone-naive prostate cancer)」と称され、(2)における前立腺癌は「ホルモン療法感受性前立腺癌(Hormone sensitive prostate cancer(HSPC)) 」と称される。
また、(3)~(5)の各ステージにおける前立腺癌は「去勢抵抗性前立腺癌(Castration resistant prostate cancer(CRPC))」と称される。去勢抵抗性前立腺癌のなかでも(3)は「化学療法剤未処置CRPC(chemo-naive CRPC)」に該当し、(5)は「化学療法剤抵抗性CRPC(chemo-failure CRPC)」に該当する。

特許文献1は、式(a)の化合物のT877A変異、L701H変異、及びH874Y変異ARに対する有効性を開示するが、他の種類の変異ARに対する有効性については開示も示唆も無い。
特許文献2は、AR拮抗薬、及びARが増悪因子となる前立腺癌、前立腺肥大症、男性化症、多毛症等の治療剤に関するが、変異AR拮抗作用に関する開示も示唆も無い。

特許文献3は、AR拮抗剤、及びARが増悪因子となる前立腺癌、前立腺肥大症、男性化症、多毛症等の治療薬に関するが、変異AR拮抗作用に関する開示も示唆も無い。

すなわち、特許文献2及び特許文献3に示される化合物の一部がAR変異を伴う前立腺癌の治療に有効であり、また別の一部がAR変異を伴う前立腺癌の治療に無効であることが見い出された。したがって、本発明は、AR変異を伴う前立腺癌の治療に有効である、当該一部の化合物においてのみに新たに見い出された、AR変異を伴う前立腺癌治療用途、特にビカルタミド耐性及びフルタミド耐性の去勢抵抗性前立腺癌(CRPC)治療用途に関する。
(1)(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩を有効成分とする、去勢抵抗性前立腺癌(CRPC)の治療用医薬組成物。
(2)去勢抵抗性前立腺癌(CRPC)がビカルタミド耐性の去勢抵抗性前立腺癌である、上記(1)の医薬組成物。
(3)去勢抵抗性前立腺癌(CRPC)がフルタミド耐性の去勢抵抗性前立腺癌である、上記(1)の医薬組成物。
(4)(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド一水和物、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩。
(5)(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド一水和物である、上記(4)の化合物又はその製薬学的に許容される塩。
(6)去勢抵抗性前立腺癌(CRPC)の治療のための、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩。
(7)去勢抵抗性前立腺癌(CRPC)の治療用医薬組成物の製造のための、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩の使用。
(8)去勢抵抗性前立腺癌(CRPC)の治療のための、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩の使用。
(9)(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩の有効量を対象に投与することからなる去勢抵抗性前立腺癌(CRPC)の治療方法。
なお、「対象」とは、その予防又は治療を必要とするヒト又はその他の動物であり、ある態様としては、その予防又は治療を必要とするヒトである。
また別の態様としては、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド一水和物である。
また別の態様としては、(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミドである。
また別の態様としては、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミドである。
また別の態様としては、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミドである。
また別の態様としては、(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミドである。
また別の態様としては、(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミドである。
また別の態様としては、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミドである。
本明細書において、「AR」はアンドロゲン受容体を意味する。
「アンドロゲン受容体変異」、「AR変異」とは、薬剤投与をはじめとする種々の要因により、アンドロゲン受容体の蛋白をコードする遺伝子の変異によるアミノ酸配列の変異が生じ、当該アンドロゲン受容体が天然アンドロゲン受容体とは異なる性質を示す現象である。「AR変異」は例えば、抗癌剤の使用による「AR変異」であり、ある態様としては、ビカルタミド又はフルタミドの使用による「AR変異」であり、別の態様としては「W741変異」又は「T877変異」であり、また別の態様としては「W741変異」であり、更に別の態様としては「W741C変異」又は「T877A変異」であり、また更に別の態様としては「W741C変異」であり、また更に別の態様としては「T877変異」であり、また更に別の態様としては「T877A変異」である。
「W741変異」とは、ビカルタミド投与患者がAWSに達した際に見い出されたアンドロゲン受容体変異の一種であり、天然AR遺伝子のコドン741に変異を呈したAR変異である。「W741変異」のある態様としては「W741C変異」又は「W741L変異」であり、別の態様としては「W741C変異」である。
「W741C変異」とは、上記「W741変異」のうち天然AR遺伝子のコドン741において、トリプトファンをコードするコドンTGGがシステインをコードするTGTに変異した、AR変異である。
「W741L変異」とは、上記「W741変異」のうち天然AR遺伝子のコドン741において、トリプトファンをコードするコドンTGGがロイシンをコードするTTGに変異した、AR変異である。
「T877変異」とは、フルタミド投与患者がAWSに達した際に呈しているアンドロゲン受容体変異の一種であり、天然AR遺伝子のコドン877に変異を呈したAR変異である。「T877変異」のある態様としては「T877A変異」である。
「T877A変異」とは、上記「T877変異」のうち、天然AR遺伝子のコドン877において、スレオニンをコードするコドンACTがアラニンをコードするコドンGCTに変異した、T877変異ARである。
「ビカルタミド耐性」とは、ビカルタミドの投与によるAWS(Anti-androgen withdrawal syndrome)が認められ、当該薬剤が無効になる前立腺癌である。ビカルタミド耐性の主な原因はW741C変異である。
「フルタミド耐性」とは、フルタミドの投与によるAWS(Anti-androgen withdrawal syndrome)が認められ、当該薬剤が無効になる前立腺癌である。フルタミド耐性の主な原因はT877A変異である。
「去勢抵抗性前立腺癌(Castration Resistant Prostate Cancer, CRPC)」とは、ホルモン療法などにより血中テストステロン値が去勢(精巣摘出)レベルに低下した状態においても病状進行を示した前立腺癌であり、chemo-naive CRPC及びchemo-failure CRPCを含む。
「Chemo-naive CRPC」とは、ドセタキセル未処置の去勢抵抗性前立腺癌である。
「Chemo-failure CRPC」とは、ドセタキセル無効例、あるいはドセタキセル処理後に病状進行を示した去勢抵抗性前立腺癌である。
更に、本発明の医薬組成物の有効成分である化合物には、これらの製薬学的に許容されるプロドラッグも含まれる。製薬学的に許容されるプロドラッグとは、加溶媒分解によりまたは生理学的条件下でアミノ基、OH、CO2H等に変換できる基を有する化合物である。プロドラッグを形成する基としては、例えば、Prog. Med., 5, 2157-2161 (1985)や「医薬品の開発」(廣川書店、1990年)第7巻 分子設計163-198に記載の基が挙げられる。
更に、本発明の医薬組成物の有効成分である化合物及びそれらの製薬学的に許容される塩には、各種の水和物や溶媒和物及び結晶多形の物質が包含される。また本発明の医薬組成物の有効成分である化合物及びそれらの製薬学的に許容される塩には、種々の放射性または非放射性同位体でラベルされた化合物も包含される。
本発明の医薬組成物の有効成分である化合物及びそれらの製薬学的に許容される塩は、その基本骨格或いは置換基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することができる。その際、官能基の種類によっては、当該官能基を原料乃至中間体の段階で適当な保護基(容易に当該官能基に転化可能な基)に置き換えておくことが製造技術上効果的な場合がある。このような官能基としては例えばアミノ基、水酸基、カルボキシル基等であり、それらの保護基としては例えば、ウッツ(P. G. M. Wuts)及びグリーン(T. W. Greene)著、「Greene's Protective Groups in Organic Synthesis (第4版、2006年)」に記載の保護基等を挙げることができ、これらを反応条件に応じて適宜選択して用いればよい。このような方法では、当該保護基を導入して反応を行った後、必要に応じて保護基を除去することにより、所望の化合物を得ることができる。
また、本発明の医薬組成物の有効成分である化合物のプロドラッグは上記保護基と同様、原料から中間体へ到る段階で特定の基を導入、或いは得られた化合物を用いてさらに反応を行うことで製造できる。反応は通常のエステル化、アミド化、脱水等、当業者により公知の方法を適用することにより行うことができる。
(一般製法)
本発明の医薬組成物の有効成分である化合物は、化合物(II)及び化合物(III)並びにC=O源となる活性化された炭酸誘導体としての化合物(IV)の反応により製造することができる。ここで適当な炭酸誘導体としては、例えば、トリホスゲン、クロロ炭酸フェニル、N,N'-カルボニルジイミダゾール(CDI)をはじめ、その他当業者に自明なものを使用することができる。
X:Cl、Br、メトキシ、エトキシ又はイソプロピルオキシ、
L:結合又はメチレン、
環A:フェニル、ピリジル、ピリミジニル、インドリル、キノリル、又はキノキサリル、
R5及びR6:同一又は互いに異なって、-CO-N(CH3)2、F、メトキシ、メチル、エチル、又はシクロプロピル、
ただし、化合物(I)は、後述の実施例化合物Ex1~22及び参考例化合物Ref1-1~1-3の何れかの化合物を意味する。
L1及びL2は脱離基を示す。)
本反応では、化合物(II)及び化合物(III)を等量若しくは一方を過剰量用い、これらの混合物を、反応に不活性な溶媒中、冷却下から加熱下、好ましくは-20℃~60℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、ベンゼン、トルエン若しくはキシレン等の芳香族炭化水素類、ジクロロメタン、1,2-ジクロロエタン(DCE)若しくはクロロホルム等のハロゲン化炭化水素類、ジエチルエーテル、テトラヒドロフラン(THF)、ジオキサン、ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド、酢酸エチル、アセトニトリル又は水、及びこれらの混合物が挙げられる。
また、本発明の医薬組成物の有効成分である化合物は、化合物(II)、及び化合物(V)を原料として製造した化合物(VI)を反応させることにより製造することができる。

本製造法における転位反応に継ぐ縮合反応は当業者によく知られた手法であり、反応溶媒、温度等は、上記製造法1と同様にして行なうことができる。具体的には後述の実施例や特許文献7の第一製法を参照することができる。

化合物(II)は、化合物(VII)と化合物(VIII)の置換反応ののち、脱保護をすることによって製造することができる。
ここで、脱離基X1の例には、ハロゲン、メタンスルホニルオキシ、p-トルエンスルホニルオキシ基等が含まれ、ある態様としてはFである。また、アミノ基の保護基Pとしては、例えばベンジル又はt-ブトキシカルボニル基である。
この反応では、化合物(VII)と化合物(VIII)とを等量若しくは一方を過剰量用い、これらの混合物を、反応に不活性な溶媒中、又は無溶媒下、冷却下から加熱還流下、好ましくは0℃から80℃において、通常0.1時間~5日間撹拌する。ここで用いられる溶媒の例としては、特に限定はされないが、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、ジメトキシエタン等のエーテル類、ジクロロメタン、1,2-ジクロロエタン、クロロホルム等のハロゲン化炭化水素類、N,N-ジメチルホルムアミド、ジメチルスルホキシド、酢酸エチル、アセトニトリル及びこれらの混合物が挙げられる。トリエチルアミン、N,N-ジイソプロピルエチルアミン若しくはN-メチルモルホリン等の有機塩基、又は炭酸カリウム、炭酸ナトリウム若しくは水酸化カリウム等の無機塩基の存在下で反応を行うのが、反応を円滑に進行させる上で有利な場合がある。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー等、通常の化学操作を適用して行なわれる。
各種の異性体は、適当な原料化合物を選択することにより製造でき、あるいは異性体間の物理化学的性質の差を利用して分離することができる。例えば、光学異性体は、ラセミ体の一般的な光学分割法(例えば、光学活性な塩基又は酸とのジアステレオマー塩に導く分別結晶化や、キラルカラム等を用いたクロマトグラフィー等)により得られ、また、適当な光学活性な原料化合物から製造することもできる。
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、あるいは静注、筋注等の注射剤、坐剤、経皮剤、経鼻剤、吸入剤等による非経口投与のいずれの形態であってもよい。投与量は症状、投与対象の年齢、性別等を考慮して個々の場合に応じて適宜決定されるが、通常、経口投与の場合、成人1日当たり0.001 mg/kg乃至100 mg/kg程度であり、これを1回で、あるいは2~4回に分けて投与する。また、症状によって静脈内投与される場合は、通常、成人1回当たり0.0001 mg/kg乃至10 mg/kgの範囲で1日に1回乃至複数回投与される。また、吸入の場合は、通常、成人1回当たり0.0001 mg/kg乃至1 mg/kgの範囲で1日に1回乃至複数回投与される。
tert-ブトキシカリウム(505 mg)及びTHF(5 ml)の混合物に氷冷下エタノール(0.264 ml)を滴下し室温で30分間撹拌した。反応液を氷冷し4-[(2S,5R)-2,5-ジメチルピペラジン-1-イル]-2-フルオロベンゾニトリル(350 mg)及びTHF(3 ml)の混合物を滴下し室温で16時間撹拌した。反応液に酢酸エチル(20 ml)及び水(10 ml)を加え分液し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧下留去した。残留物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0-96/4)で精製して4-[(2S,5R)-2,5-ジメチルピペラジン-1-イル]-2-エトキシベンゾニトリル315 mg(81%)を得た。ESI+:260
製造例2
ピペラジン-2-オン(5.06 g)及びDMF(50 ml)の混合物に、トリエチルアミン(8 ml)及び2-クロロ-4-フルオロベンゾニトリル(8.1 g)を加え80℃で1日撹拌した。反応液に水を加え析出した固体をろ取し、水及びジイソプロピルエーテルで洗浄して2-クロロ-4-(3-オキソピペラジン-1-イル)ベンゾニトリル11.2 g(94%)を得た。EI+:235
製造例3
2-クロロ-4-(3-オキソピペラジン-1-イル)ベンゾニトリル(11.2 g)及びDMF(160 ml)の混合物に、水素化ナトリウム(2.2 g、55%流動パラフィン分散体)を加え室温で10分間撹拌した後に臭化ベンジル(6 ml)を加え室温で1時間撹拌した。反応液に水を加え析出した固体をろ取し、水及びジイソプロピルエーテルで洗浄して4-(4-ベンジル-3-オキソピペラジン-1-イル)-2-クロロベンゾニトリル12.74 g(82%)を得た。EI+:325
4-(4-ベンジル-3-オキソピペラジン-1-イル)-2-クロロベンゾニトリル(12.7 g)、ヨウ化メチル(7 ml)及びTHF(100 ml)の混合物を-78℃に冷却した。別途、ジイソプロピルアミン(16 ml)及びTHF(100 ml)の混合物に-78℃でn-ブチルリチウム(40 ml、2.77Mヘキサン溶液)を滴下してリチウムジイソプロピルアミド(LDA)溶液を調整した。LDA溶液を先の混液にカニューレで40分かけて加え更に2時間撹拌した。反応液に少量の水を加えて室温に戻し、水及び酢酸エチルを加えて分液操作を行った。水層に酢酸エチルを加えて分液操作を行った。有機層を合わせて飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧下留去した。残留物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=10/0-5/5)で精製して4-(4-ベンジル-2,2-ジメチル-3-オキソピペラジン-1-イル)-2-クロロベンゾニトリルを6.14 g(44%)得た。EI+:353
製造例5
4-(4-ベンジル-2,2-ジメチル-3-オキソピペラジン-1-イル)-2-クロロベンゾニトリル(6.13 g)及びTHF(10 ml)の混合物に1,1,3,3-テトラメチルジシロキサン(9.12 g)及びヘキサクロロ白金(IV)酸六水和物(0.8 g)を加え50℃で2日間撹拌した。反応液の不溶物を除いた後に溶媒を減圧下留去し、残留物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=10/0-5/5)で2回精製して4-(4-ベンジル-2,2-ジメチルピペラジン-1-イル)-2-クロロベンゾニトリル1.87 g(32%)を得た。EI+:339
製造例6
4-(4-ベンジル-2,2-ジメチルピペラジン-1-イル)-2-クロロベンゾニトリル(1.87 g)及びDCE(20 ml)の混合物にクロロギ酸1-クロロエチル(1 ml)を加え加熱還流下で1時間攪拌した。反応液を放冷後、溶媒を減圧下留去し、残留物にメタノール(20 ml)を加え加熱還流下で更に1時間撹拌した。反応液を放冷して減圧下濃縮し、水及び少量の1M塩酸を加えた。水溶液を酢酸エチルで洗浄し、水層を1M水酸化ナトリウム水溶液で塩基性とした。酢酸エチルを加えて分液操作を行なった。有機層を飽和食塩水で洗浄し、水硫酸ナトリウムで乾燥後、溶媒を減圧下留去して2-クロロ-4-(2,2-ジメチルピペラジン-1-イル)ベンゾニトリル1.31 g(95%)を得た。EI+:250
ビス(トリクロロメチル)カーボネート(178 mg)及びTHF(6 ml)の混合物に、氷冷下2-クロロ-4-[(2S,5R)-2,5-ジメチルピペラジン-1-イル]ベンゾニトリル(375 mg)、トリエチルアミン(0.314 ml)及びTHF(3 ml)の混合物を加え室温で1時間撹拌した後に反応液を氷冷し、4-(アミノメチル)安息香酸メチル(273 mg)、トリエチルアミン(0.23 ml)及びTHF (3 ml)の混合物を加え室温で更に3時間撹拌した。反応液に飽和炭酸水素ナトリウム水溶液(10 ml)、水(15 ml)及び酢酸エチル(15 ml)を加え分液操作を行い、有機層を無水硫酸ナトリウムで乾燥後溶媒を減圧下留去した。残留物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0-98/2)で精製して、4-[({[(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチルピペラジン-1-イル]カルボニル}アミノ)メチル]安息香酸メチル582 mg(88%)を得た。EI+:441
製造例8
4-[({[(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチルピペラジン-1-イル]カルボニル}アミノ)メチル]安息香酸メチル(582 mg)及びメタノール/THF(1/1, 6 ml)の混合物に1M水酸化ナトリウム水溶液(3.3 ml)を加え室温で14時間撹拌した。反応液を減圧下濃縮し氷冷下1M塩酸(3.3 ml)及び水(10 ml)を加え析出した固体をろ取して4-[({[(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチルピペラジン-1-イル]カルボニル}アミノ)メチル]安息香酸512 mg(91%)を得た。EI+:428
4-[({[(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチルピペラジン-1-イル]カルボニル}アミノ)メチル]安息香酸(256 mg)及びDMF(3 ml)の混合物に、氷冷下ジメチルアミン塩酸塩(147 mg)、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩(WSC・HCl)(173 mg)、1-ヒドロキシ-1H-ベンゾトリアゾール(HOBt)(41 mg)及びトリエチルアミン(0.251 ml)を加え室温で18時間撹拌した。反応液に水(10 ml)及び酢酸エチル(10 ml)を加え分液操作を行なった。再度水層に酢酸エチルを加え分液操作を行なった。合わせた有機層を飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧下留去した。残留物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0-96/4)で精製して、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-[4-(ジメチルカルバモイル)ベンジル]-2,5-ジメチルピペラジン-1-カルボキサミド160 mg(59%)を得た。
2-メトキシピリミジン-5-アミン(120 mg)及びピリジン(4 ml)の混合物に氷冷下、クロロ炭酸フェニル(0.132 ml)を加え室温で1.5時間撹拌した。次いで、2-クロロ-4-[(2S,5R)-2,5-ジメチルピペラジン-1-イル]ベンゾニトリル(200 mg)及びピリジン(3 ml)の混合物を加え100℃で1時間撹拌した。反応液を放冷後、減圧下濃縮し、残渣にトルエン(15 ml x 2)を加え共沸した。残渣に酢酸エチル(20 ml)を加え、飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄した。更に無水硫酸ナトリウムで乾燥後、溶媒を減圧下留去した。残留物をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/0-96/4)で精製して、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド258 mg(81%)を得た。
実施例16
2-クロロ-6-メチルイソニコチン酸(151 mg)及び酢酸エチル(3 ml)の混合物にトリエチルアミン(0.184 ml)及びDPPA(0.208 ml)を加え、室温で2.5時間撹拌した。反応液にトルエン(15 ml)を加え飽和炭酸水素ナトリウム水溶液で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧下留去した。残留物をトルエン(5 ml)に溶解し、加熱還流下で40分間撹拌した後に室温まで放冷し、2-クロロ-4-[(2S,5R)-2,5-ジメチルピペラジン-1-イル]ベンゾニトリル(200 mg)及びアセトニトリル(3 ml)の混液を加え、室温で1時間撹拌した。析出した固体をろ取してアセトニトリルで洗浄し、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド235 mg(70%)得た。
実施例22
特許文献3の実施例3-9の化合物である、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド(化合物A)(250 mg)を室温で50%アセトニトリル水(6 ml)で結晶化して (2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド一水和物の結晶 (111 mg)(44%)を得た。
参考例1-3
2-クロロ-4-[(2S,5R)-2,5-ジメチルピペラジン-1-イル]ベンゾニトリル(100 mg)、メチルキノキサリン-2-イルカルバメート(72mg)及びトルエン(2ml)の懸濁液にアルミナ(80mg)を加え加熱還流下で1日攪拌した。反応液を放冷した後に減圧下濃縮し、残留物をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール = 100/0-95/5)で精製した。得られた精製物に酢酸エチル及びジイソプロピルエーテルを加え、(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-(キノキサリン-2-イル)ピペラジン-1-カルボキサミド34mg(23%)を固体として得た。





試験例1:ヒトW741C及びT877A変異AR転写活性化阻害作用
CHO-K1細胞にW741C或いはT877A変異AR発現ベクター(pSG5-W741C-hAR或いはpcDNA3.1-T877A-hAR)をトランスフェクトすることにより、ヒトW741CおよびT877A変異AR安定発現細胞を取得した。また、これらの細胞に、ARが活性化したときにルシフェラーゼを発現するように、ルシフェラーゼレポーターベクターもトランスフェクトし、ヒトW741C及びT877A変異AR安定発現細胞を取得した。
W741C或いはT877A変異AR安定発現CHO-K1細胞を、それぞれ96well細胞培養用ルミノプレートに2×104個播き、37℃で一晩インキュベートした後、DHT(終濃度 0.3 nM)と同時に種々濃度の被検化合物を添加した。その後更に、37℃で一晩インキュベートした後、ルシフェラーゼアッセイシステムで処理した細胞の発光量を、ルミノメーターを用いて測定し、W741C或いはT877A変異AR転写活性化によるルシフェラーゼ活性とした。
本発明の医薬組成物の有効成分である化合物の転写活性化阻害作用をルシフェラーゼ阻害活性として評価した。IC50値はSigmoid-Emaxモデル非線形回帰分析によって算出した。
上記試験の被検化合物として、ビカルタミド(Bicalutamide)、ヒドロキシフルタミド(Hydroxyflutamide)、本発明の有効成分である化合物(化合物A(Compound A、特許文献3、実施例3-9)、実施例7, 8, 11, 16, 18, 19, 及び22の化合物)、並びに参考例1-1~1-3を用いて、野生型AR(Wild)、W741C変異AR及びT877A変異ARのそれぞれの受容体に対する転写活性化阻害作用を評価した。各被検化合物の受容体阻害作用をIC50値として算出し下記表にその値を示す。なお、「-」は未実施を意味する。
表中のIC50値が>1000 nMは、転写活性化阻害作用が見い出されずAR拮抗活性がほぼ無効であることを意味する。
ビカルタミドは既存AR拮抗薬であり、ビカルタミド耐性の特徴であるW741C変異を有する転写活性化阻害作用が見い出されずW741C変異AR受容体に対して拮抗活性がほぼ無効であることが確認された。
ヒドロキシフルタミドは、フルタミドの活性代謝物であり、フルタミド耐性の特徴であるA877A変異を有するAR受容体に対して拮抗活性がほぼ無効であることが確認された。
本発明の医薬組成物の有効成分である化合物である、化合物A(Compound A)、実施例7、8、11、16、18、19及び22の化合物は野生型AR、W741C変異AR、及びA877A変異ARの何れのARに対しても転写活性化阻害作用が認められ、AR拮抗活性を有することが確認された。
参考例1-1、1-2、及び1-3の化合物は、野生型ARに対しては転写活性化阻害作用が認められ野生型AR拮抗活性が認められるが、化合物A(Compound A)、実施例7、8、11、16、18、19、及び22の化合物と比較して、W741C又はT877A変異AR、或いはその両方に対する転写活性化阻害作用が大きく減弱され、ゆえに変異AR拮抗活性が大きく減弱されることが確認された。
なお、実施例22(Ex 22)は、化合物Aの一水和物であり、特許文献3には開示も示唆もない。
KUCaPは、京都大学で樹立されたヒト前立腺癌細胞であり(Cancer Res 2005; 65: 9611-9616)、W741C変異ARを有し、in vivo(マウス)での継代が可能な細胞である。本細胞片を雄性SCIDマウスの背部皮下に移植し、腫瘍体積が約200~400 mm3になった時点で、腫瘍体積が各群で均等になるように群分け(1群5匹)をし、被験化合物の投与を開始した。被験化合物は、25%プロピレングリコール/25%ツイーン80/50% 精製水に溶解し、5 mg/kg/10 mLで1日2回(10 mg/kg/day)14日間経口投与した。なお、対照群には、25%プロピレングリコール/25%ツイーン80/50%精製水を5 mL/kgの容量で投与した。腫瘍径はノギスを用いて、腫瘍の長径(mm)と短径(mm)を測定し、「長径×短径2×0.5」の計算式により、腫瘍体積(mm3)を算出した。また、被験薬の阻害率[%]は、以下の計算式により算出した。阻害率[%]=100×{1―〔(被験化合物群の14日目の腫瘍体積-被験化合物群の投与開始日の腫瘍体積)〕/〔(対照群の14日目の腫瘍体積-対照群の投与開始日の腫瘍体積)〕}
Yoshida T, Kinoshita H, Segawa T et al. "Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from bicalutamide-treated patient" Cancer Res 2005; 65: 9611-9616.

CHO-K1細胞にW741C或いはT877A変異AR発現ベクター(pSG5-W741C-hAR或いはpcDNA3.1-T877A-hAR)をトランスフェクトすることにより、ヒトW741C及びT877A変異AR一過性強制発現細胞を取得した。本細胞を24 wellプレートに5×104 cell/wellで播種し、37℃で一晩培養した。培地を除去し、DCC-FBSを添加した培地で希釈した被検化合物および[3H]DHTを添加し、37℃で4時間培養した。培地を除去した後、lysis bufferを加えて細胞を溶解し、上清の放射活性を測定した。この放射活性値から、[3H]DHTの特異的結合に対する被検化合物の阻害活性のIC50を求めた。
1) W741C-hAR-LNCaPの取得
ヒト前立腺癌細胞株であるLNCaP細胞にW741C変異AR発現ベクター(pSG5-W741C-hAR)をトランスフェクトすることにより、W741C-hAR-LNCaPを取得した。
2) W741C-hAR-LNCaP増殖促進作用
Poly-L-lysineコートされた96 wellプレートにW741C-hAR-LNCaP細胞を5×103 cell/wellで播種して1日培養し、DCC-FBSを含む培地で希釈した化合物、DHT或いは溶媒(DMSO)を添加し更に培養した。7日後にスルホローダミンB法(後述の文献参照)を用いて各wellの蛋白質量を測定した。溶媒のみ(被検化合物なし)のwellの蛋白質量と比較して、それ以上の蛋白質量増加を化合物の増殖促進作用として評価した。
3) W741C-hAR-LNCaP増殖阻害作用
Poly-L-lysineコートされた96 well プレートにW741C-hAR-LNCaP細胞を5×103 cell/wellで播種して1日培養し、DCC-FBSを含む培地で希釈した被検化合物(或いは溶媒)およびDHT(1 nM)両方を添加し更に培養した。7日後にスルホローダミンB法を用いて各wellの蛋白質量を測定した。阻害率(%)は、以下の式により算出した。
阻害率(%)= 100〔(I―B)―(X―B)〕/(I―B)
I:1 nM DHTのみを添加した場合の蛋白質量
B:溶媒のみ添加した場合の蛋白質量
X:本化合物と1 nM DHTを同時に添加した場合の蛋白質量
参考文献1:Skehan P, Storeng R, Scudiero D et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82: 1107-1112.
参考文献2:Papazisis KT, Geromichalos GD, Dimitriadis KA et al. Optimization of the sulforhodamine B colorimetric assay. J Immunol Methods 1997; 208: 151-158.
Claims (9)
- (2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩を有効成分とする、去勢抵抗性前立腺癌の治療用医薬組成物。 - 去勢抵抗性前立腺癌がビカルタミド耐性の去勢抵抗性前立腺癌である、請求項1の医薬組成物。
- 去勢抵抗性前立腺癌がフルタミド耐性の去勢抵抗性前立腺癌である、請求項1の医薬組成物。
- (2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド一水和物、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩。 - (2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド一水和物である、請求項4の化合物又はその製薬学的に許容される塩。
- 去勢抵抗性前立腺癌の治療のための、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩。 - 去勢抵抗性前立腺癌の治療用医薬組成物の製造のための
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩の使用。 - 去勢抵抗性前立腺癌の治療のための、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩の使用。 - (2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-シクロプロピルピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(4-シアノ-3-メトキシフェニル)-N-(2,6-ジメチルピリミジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-クロロ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシピリミジン-5-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、
(2R,5S)-4-(3-ブロモ-4-シアノフェニル)-N-(2-メトキシ-6-メチルピリジン-4-イル)-2,5-ジメチルピペラジン-1-カルボキサミド、及び
(2R,5S)-4-(3-クロロ-4-シアノフェニル)-2,5-ジメチル-N-[2-(ピペリジン-1-イル)ピリミジン-5-イル]ピペラジン-1-カルボキサミド、
並びにそれらの製薬学的に許容される塩からなる群より選択される化合物又はその製薬学的に許容される塩の有効量を対象に投与することからなる去勢抵抗性前立腺癌の治療方法。
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2813063A CA2813063A1 (en) | 2010-10-22 | 2011-10-21 | Antagonist for mutated androgen receptor |
| EP11834469.6A EP2631233A4 (en) | 2010-10-22 | 2011-10-21 | ANTAGONIST FOR A MUTANT ANDROGEN RECEPTOR |
| JP2012539778A JPWO2012053630A1 (ja) | 2010-10-22 | 2011-10-21 | 変異アンドロゲン受容体拮抗薬 |
| AU2011318875A AU2011318875A1 (en) | 2010-10-22 | 2011-10-21 | Antagonist for mutated androgen receptor |
| KR1020137011626A KR20130139979A (ko) | 2010-10-22 | 2011-10-21 | 변이 안드로겐 수용체 길항약 |
| BR112013009274-2A BR112013009274A2 (ja) | 2010-10-22 | 2011-10-21 | Variation androgen receptor antagonist |
| MX2013004517A MX2013004517A (es) | 2010-10-22 | 2011-10-21 | Antagonistas para el receptor androgeno mutado. |
| CN2011800505477A CN103180309A (zh) | 2010-10-22 | 2011-10-21 | 突变雄激素受体拮抗剂 |
| US13/877,358 US20130197009A1 (en) | 2010-10-22 | 2011-10-21 | Antagonist for mutant androgen receptor |
| EA201390598A EA201390598A1 (ru) | 2010-10-22 | 2011-10-21 | Антагонист мутантного андрогенного рецептора |
| IL225395A IL225395A0 (en) | 2010-10-22 | 2013-03-21 | A mutated androgen receptor antagonist |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010237242 | 2010-10-22 | ||
| JP2010-237242 | 2010-10-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012053630A1 true WO2012053630A1 (ja) | 2012-04-26 |
Family
ID=45975338
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/074261 WO2012053630A1 (ja) | 2010-10-22 | 2011-10-21 | 変異アンドロゲン受容体拮抗薬 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20130197009A1 (ja) |
| EP (1) | EP2631233A4 (ja) |
| JP (1) | JPWO2012053630A1 (ja) |
| KR (1) | KR20130139979A (ja) |
| CN (1) | CN103180309A (ja) |
| AU (1) | AU2011318875A1 (ja) |
| BR (1) | BR112013009274A2 (ja) |
| CA (1) | CA2813063A1 (ja) |
| EA (1) | EA201390598A1 (ja) |
| IL (1) | IL225395A0 (ja) |
| MX (1) | MX2013004517A (ja) |
| TW (1) | TW201305130A (ja) |
| WO (1) | WO2012053630A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013058361A1 (ja) | 2011-10-21 | 2013-04-25 | アステラス製薬株式会社 | アンドロゲン受容体拮抗化合物の結晶 |
| WO2014066864A2 (en) * | 2012-10-26 | 2014-05-01 | Memorial Sloan-Kettering Cancer Center | Androgen receptor variants and methods for making and using |
| JP2016516005A (ja) * | 2013-02-25 | 2016-06-02 | ノバルティス アーゲー | 新規のアンドロゲンレセプター変異 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9682960B2 (en) | 2013-12-19 | 2017-06-20 | Endorecherche, Inc. | Non-steroidal antiandrogens and selective androgen receptor modulators with a pyridyl moiety |
| CN114761003B (zh) * | 2019-09-23 | 2023-12-29 | 冰洲石生物科技公司 | 具有雄激素受体降解活性的新型脲类及其用途 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000017163A1 (fr) | 1998-09-22 | 2000-03-30 | Yamanouchi Pharmaceutical Co., Ltd. | Derives de cyanophenyle |
| JP2001328938A (ja) * | 2000-03-17 | 2001-11-27 | Yamanouchi Pharmaceut Co Ltd | シアノフェニル誘導体を有効成分とする医薬 |
| WO2004007471A1 (ja) | 2002-07-12 | 2004-01-22 | Yamanouchi Pharmaceutical Co., Ltd. | N−フェニル−(2r,5s)ジメチルピペラジン誘導体 |
| WO2005040136A1 (en) | 2003-10-21 | 2005-05-06 | Bristol-Myers Squibb Company | Piperazine derivatives and their use as modulators of nuclear hormone receptor function |
| US20050159468A1 (en) | 2003-05-09 | 2005-07-21 | Arwed Cleve | Anti-androgenic pyrrolidines with tumor-inhibiting action |
| WO2006064944A1 (en) | 2004-12-14 | 2006-06-22 | Takeda Pharmaceutical Company Limited | Substituted pyrrole derivative |
| WO2009003077A1 (en) | 2007-06-27 | 2008-12-31 | Bristol-Myers Squibb Company | Fused heterocyclic compounds useful as modulators of nuclear hormone receptor function |
| WO2009028543A1 (ja) | 2007-08-30 | 2009-03-05 | Takeda Pharmaceutical Company Limited | 置換ピラゾール誘導体 |
| WO2009059077A1 (en) | 2007-11-01 | 2009-05-07 | Bristol-Myers Squibb Company | Fused heterocyclic compounds useful as modulators of nuclear hormone receptor function |
-
2011
- 2011-10-21 BR BR112013009274-2A patent/BR112013009274A2/ja not_active IP Right Cessation
- 2011-10-21 EP EP11834469.6A patent/EP2631233A4/en not_active Withdrawn
- 2011-10-21 KR KR1020137011626A patent/KR20130139979A/ko not_active Application Discontinuation
- 2011-10-21 TW TW100138310A patent/TW201305130A/zh unknown
- 2011-10-21 MX MX2013004517A patent/MX2013004517A/es unknown
- 2011-10-21 CN CN2011800505477A patent/CN103180309A/zh active Pending
- 2011-10-21 CA CA2813063A patent/CA2813063A1/en not_active Abandoned
- 2011-10-21 JP JP2012539778A patent/JPWO2012053630A1/ja active Pending
- 2011-10-21 AU AU2011318875A patent/AU2011318875A1/en not_active Abandoned
- 2011-10-21 US US13/877,358 patent/US20130197009A1/en not_active Abandoned
- 2011-10-21 WO PCT/JP2011/074261 patent/WO2012053630A1/ja active Application Filing
- 2011-10-21 EA EA201390598A patent/EA201390598A1/ru unknown
-
2013
- 2013-03-21 IL IL225395A patent/IL225395A0/en unknown
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000017163A1 (fr) | 1998-09-22 | 2000-03-30 | Yamanouchi Pharmaceutical Co., Ltd. | Derives de cyanophenyle |
| JP2001328938A (ja) * | 2000-03-17 | 2001-11-27 | Yamanouchi Pharmaceut Co Ltd | シアノフェニル誘導体を有効成分とする医薬 |
| WO2004007471A1 (ja) | 2002-07-12 | 2004-01-22 | Yamanouchi Pharmaceutical Co., Ltd. | N−フェニル−(2r,5s)ジメチルピペラジン誘導体 |
| US20050159468A1 (en) | 2003-05-09 | 2005-07-21 | Arwed Cleve | Anti-androgenic pyrrolidines with tumor-inhibiting action |
| WO2005040136A1 (en) | 2003-10-21 | 2005-05-06 | Bristol-Myers Squibb Company | Piperazine derivatives and their use as modulators of nuclear hormone receptor function |
| WO2006064944A1 (en) | 2004-12-14 | 2006-06-22 | Takeda Pharmaceutical Company Limited | Substituted pyrrole derivative |
| WO2009003077A1 (en) | 2007-06-27 | 2008-12-31 | Bristol-Myers Squibb Company | Fused heterocyclic compounds useful as modulators of nuclear hormone receptor function |
| WO2009028543A1 (ja) | 2007-08-30 | 2009-03-05 | Takeda Pharmaceutical Company Limited | 置換ピラゾール誘導体 |
| WO2009059077A1 (en) | 2007-11-01 | 2009-05-07 | Bristol-Myers Squibb Company | Fused heterocyclic compounds useful as modulators of nuclear hormone receptor function |
Non-Patent Citations (14)
| Title |
|---|
| "Bunshi Sekkei (Drug Design", vol. 7, 1990, HIROKAWA PUBLISHING COMPANY, article "Iyakuhin no Kaihatsu (Pharmaceutical research and development", pages: 163 - 198 |
| CANCER RES, vol. 65, 2005, pages 9611 - 9616 |
| CANCER RESEARCH, vol. 62, 2002, pages 1496 - 1502 |
| CANCER RESEARCH, vol. 63, 2003, pages 149 - 153 |
| J. CELL. BIOCHEM, vol. 91, 2007, pages 3 - 12 |
| JOURNAL OF CELLULAR BIOCHEMISTRY, vol. 91, 2004, pages 3 - 12 |
| MIYAMOTO H. ET AL.: "Molecular Basis for the Antiandrogen Withdrawal Syndrome", JOUNAL OF CELLULAR BIOCHEMISTRY, vol. 91, 2004, pages 3 - 12, XP055083012 * |
| PAPAZISIS KT; GEROMICHALOS GD; DIMITRIADIS KA ET AL.: "Optimization of the sulforhodamine B colorimetric assay", J IMMUNOL METHODS, vol. 208, 1997, pages 151 - 158, XP004101640, DOI: doi:10.1016/S0022-1759(97)00137-3 |
| PROG. MED., vol. 5, 1985, pages 2157 - 2161 |
| SCIENCE, vol. 324, 2009, pages 787 - 790 |
| See also references of EP2631233A4 |
| SKEHAN P; STORENG R; SCUDIERO D ET AL.: "New colorimetric cytotoxicity assay for anticancer-drug screening", J NATL CANCER INST, vol. 82, 1990, pages 1107 - 1112, XP001022779, DOI: doi:10.1093/jnci/82.13.1107 |
| WUTS; GREENE: "Greene's Protective Groups in Organic Synthesis", 2006 |
| YOSHIDA T; KINOSHITA H; SEGAWA T ET AL.: "Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from bicalutamide-treated patient", CANCR RES, vol. 65, 2005, pages 9611 - 9616 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013058361A1 (ja) | 2011-10-21 | 2013-04-25 | アステラス製薬株式会社 | アンドロゲン受容体拮抗化合物の結晶 |
| US20140275105A1 (en) * | 2011-10-21 | 2014-09-18 | Astellas Pharma Inc. | Crystal of androgen receptor antagonistic compound |
| WO2014066864A2 (en) * | 2012-10-26 | 2014-05-01 | Memorial Sloan-Kettering Cancer Center | Androgen receptor variants and methods for making and using |
| WO2014066864A3 (en) * | 2012-10-26 | 2014-08-28 | Memorial Sloan-Kettering Cancer Center | Androgen receptor variants and methods for making and using |
| AU2013334075B2 (en) * | 2012-10-26 | 2018-11-29 | Memorial Sloan-Kettering Cancer Center | Androgen receptor variants and methods for making and using |
| US10206911B2 (en) | 2012-10-26 | 2019-02-19 | Memorial Sloan-Kettering Cancer Center | Androgen receptor variants and methods for making and using |
| JP2016516005A (ja) * | 2013-02-25 | 2016-06-02 | ノバルティス アーゲー | 新規のアンドロゲンレセプター変異 |
Also Published As
| Publication number | Publication date |
|---|---|
| EA201390598A1 (ru) | 2013-08-30 |
| EP2631233A1 (en) | 2013-08-28 |
| CA2813063A1 (en) | 2012-04-26 |
| AU2011318875A1 (en) | 2013-05-02 |
| MX2013004517A (es) | 2013-06-03 |
| TW201305130A (zh) | 2013-02-01 |
| EP2631233A4 (en) | 2014-03-19 |
| KR20130139979A (ko) | 2013-12-23 |
| JPWO2012053630A1 (ja) | 2014-02-24 |
| IL225395A0 (en) | 2013-06-27 |
| CN103180309A (zh) | 2013-06-26 |
| BR112013009274A2 (ja) | 2018-05-02 |
| US20130197009A1 (en) | 2013-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11873283B2 (en) | Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewith | |
| CN107011242B (zh) | 用于治疗尤文肉瘤家族肿瘤的方法和组合物 | |
| RU2434851C1 (ru) | Циклические n, n'-диарилтиомочевины или n, n'-диарилмочевины - антагонисты андрогенных рецепторов, противораковое средство, способ получения и применения | |
| EP2864289B1 (en) | Cyclopropanecarboxamido-substituted aromatic compounds as anti-tumor agents | |
| WO2012053606A1 (ja) | アリールアミノヘテロ環カルボキサミド化合物 | |
| JP2007055940A (ja) | ピラゾロピリミジン誘導体 | |
| US20040214880A1 (en) | Oxime derivatives and their use as pharmaceutically active agents | |
| JP2019504830A (ja) | Egfr阻害剤としての新規フッ素化キナゾリン誘導体 | |
| AU2013283487B2 (en) | A phenyl triazole derivative and its use for modulating the GABAA receptor complex | |
| JP6581193B2 (ja) | 置換2−チオキソ−イミダゾリジン−4−オン及びそのスピロ類似体、抗癌有効成分、医薬組成物、医薬製剤、並びに前立腺癌の治療法 | |
| WO2012053630A1 (ja) | 変異アンドロゲン受容体拮抗薬 | |
| EP1716867A1 (en) | Preventives for migraine | |
| US11560371B2 (en) | Cereblon binding compounds, compositions thereof, and methods of treatment therewith | |
| US20080171788A1 (en) | Medicament For Irritable Bowel Syndrome | |
| WO2012115118A1 (ja) | ポリペプチド化合物 | |
| CN112759545B (zh) | 3-(二甲氨基甲基)哌啶-4-醇类衍生物及其制备方法和药物用途 | |
| JP2002510625A (ja) | Mrp1の阻害方法 | |
| US20080221116A1 (en) | ISOXAZOLINE ALPHA 1a/1d ADRENORECEPTOR ANTAGONISTS | |
| JP2007269628A (ja) | 医薬化合物の結晶 | |
| US20220267268A1 (en) | New tricyclic 5-ht2 antagonists | |
| JP5220611B2 (ja) | 1,4−ベンゾジアゼピン誘導体、それを含有する医薬組成物およびそれらの医薬用途 | |
| CN116693446A (zh) | 苯基脲类衍生物及其医药用途 | |
| JP2004511486A (ja) | デルタ−オピオイドアゴニストおよびアンタゴニストとしてのモルフィノイド誘導体 | |
| CN117561249A (zh) | Cereblon结合化合物、其组合物及其用于治疗的方法 | |
| WO2008118092A1 (en) | Maleate salt of 3-bromo-n-{ (2s) -2- (4-f luorophenyl) -4- [3- (4-acetylpiperazin-1-yl) azetidin-1-yl] butyl} -methyl-5- (trifluoromethyl)benzamide for the treatment of gastrointestinal disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11834469 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012539778 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 225395 Country of ref document: IL Ref document number: 12013500534 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2813063 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13877358 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011834469 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2013/004517 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2011318875 Country of ref document: AU Date of ref document: 20111021 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20137011626 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201306311 Country of ref document: UA Ref document number: 201390598 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013009274 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013009274 Country of ref document: BR Kind code of ref document: A2 Effective date: 20130416 |